 Recently, two key pieces of guidance were released from Food and Drug Administration (FDA) and European Medicines Agency (EMA) regarding risk based approaches to clinical research. These documents include FDA’s “Guidance for Industry: Oversight of Clinical Investigations—A Risk-Based Approach to Monitoring” and EMA’s “Reflection paper on risk based quality management in clinical trials”.
Recently, two key pieces of guidance were released from Food and Drug Administration (FDA) and European Medicines Agency (EMA) regarding risk based approaches to clinical research. These documents include FDA’s “Guidance for Industry: Oversight of Clinical Investigations—A Risk-Based Approach to Monitoring” and EMA’s “Reflection paper on risk based quality management in clinical trials”. Risk Management Process

- What might go wrong?
- What is the likelihood (probability) it will go wrong?
- What are the chances we will discover (detectability) the issue?
- What are the consequences (severity)?
- What is the acceptable level of risk for the clinical study?
- Is the risk above an acceptable level?
- What can be done to reduce or eliminate risks?
- Are new risks introduced as a result of the identified risks being controlled
- How complex is the study design? Is it an adaptive design trial?
- Does the study have any interpretive or subjective data endpoints?
- Does the study population include a vulnerable population or subset?
- Are sites located in a region of the world there are differences in the standards of medical practice and/or infrastructure of clinical research practice?
- What is the experience of the clinical investigator? What is the sponsor’s experience working with the clinical investigator?
- Is the site using electronic data capture (EDC) systems?
- Does the investigational product have any safety concerns?
- What is the stage of the study? Is the study in the enrollment stage or the follow-up stage?
After asking and answering the questions above, risks are identified. In clinical research, studies are dependent on so many factors and subsequently the control tactics for each risk are going to be project, sponsor, and site dependent. The following two examples illustrate how risk management can be applied to issues commonly observed within clinical trials for each phase of the clinical project. To explore the listed examples in more detail, please download our whitepaper.

Example 2, Training of Clinical Research Sites with Varying Experience. This example examines a multi-year clinical study that has two sites. In the first year of the study, it is noted that Site A has little experience and Site B has many years of experience conducting clinical studies.
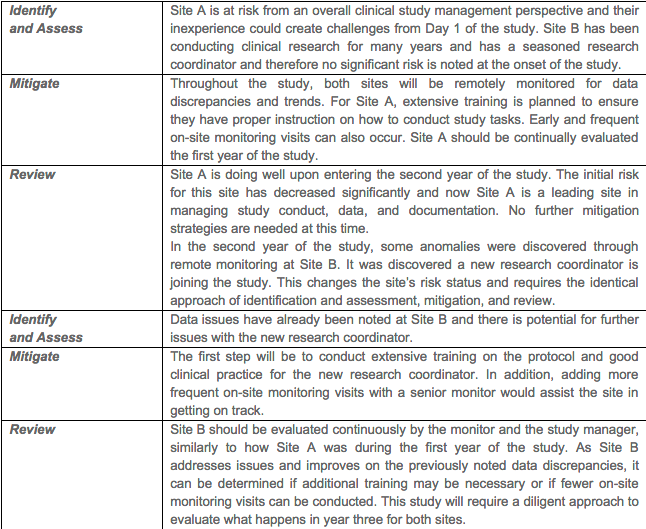 In summary, risk management strategies can and should be applied to the clinical research industry. FDA and EMA have released guidance documents that discuss and reference the incorporation of these principles into the clinical trial development, execution, and closure. The advantage of incorporation of these quality principles is an investment into the overall success of the clinical trial and will save time, resources, and likely eliminate, prevent, and/or minimize subject safety and data integrity risks.
In summary, risk management strategies can and should be applied to the clinical research industry. FDA and EMA have released guidance documents that discuss and reference the incorporation of these principles into the clinical trial development, execution, and closure. The advantage of incorporation of these quality principles is an investment into the overall success of the clinical trial and will save time, resources, and likely eliminate, prevent, and/or minimize subject safety and data integrity risks.
Note: This article is used with permission from GxP Lifeline and has been previously published at www.mastercontrol.com.”
Bio:
Emily Haglund, MS, CCRP is a Clinical Auditor for IMARC Research, Inc. IMARC is a medical device CRO, specializing in monitoring, auditing, training and consulting services. Along with conducting quality assurance activities and audits, Emily had worked with clinical teams in the medical device industry and healthcare policy research. She is a member of the Society for Quality Assurance (SQA) and the Society for Clinical Research Associates (SoCRA). Contact her at ehaglund@imarcresearch.com.