FDA classically has defined the requirements for validation under 21 CFR 820 and 210/211 regulations as a comprehensive testing process where all systems are given thorough examination and tested under equal weight, complete with an exhaustive evaluation process.
Recent guidance and initiatives by FDA (Process Validation: General Principles and Practices) and ICH (Q11: DEVELOPMENT AND MANUFACTURE OF DRUG SUBSTANCES) have provided a streamlined risk based approach under an updated life cycle management methodology. Under this scenario, a new definition of validation has emerged, best described by FDA as;
“the collection and evaluation of data, from the process design stage through production, which establishes scientific evidence that a process is capable of consistently delivering quality products.”
This is in contrast to the classical definition as perhaps best emphasized in the device regulations under 21 CFR 820.75:
“Where the results of a process cannot be fully verified by subsequent inspection and test, the process shall be validated with a high degree of assurance and approved according to established procedures.”
What this means is that a risk-based life cycle management approach with relevant scientific rationale and evidence can be used in lieu of a traditional top-down comprehensive approach. Many of us remember the golden rule of validation; triplicate test runs as an output of this classic approach. It didn’t matter the complexity or simplicity of the system, just always apply the test in triplicate.
Essentially what FDA and ICH are now saying is that if you can justify a different test plan with a risk-based approach, it is fine with them. The end result is that many validation processes can be streamlined and production delays eliminated. Below is essentially a discussion on the ‘best practices” approach I have successfully used to meet these updated FDA and ICH guidances.
USER REQUIREMENT SPECIFICATIONS
Whether the intended validation effort is for equipment, processes or software, it is highly recommended that a User Requirement Speciation (URS) be written. This facilitates a starting point and traceability to ensure that basic functions are established. These basic functions will be used later for assessing risks. Software validation typically also has a Functional Requirement Specifications (FRS) that follows the URS in a logical, traceable way. The FRS shows the way the software post-configuration will meet the requirements of the URS.
RISK ASSESSMENT
A risk assessment follows the URS/FRS process. Utilizing the risk management methodologies of ISO 14971, it is imperative to establish the acceptance criteria and risk levels before applying a risk assessment to the functional processes developed in the URS/FRS. The best practices approach developed below is a three level system with low (green) medium (yellow) and high (red) risk categories. These can be characterized as follows:
- High: Failure would severely affect safety and quality processes.
- Medium: Failure would have a moderate impact on safety and quality processes.
- Low: Failure would have a minor impact on patient safety or product quality.
From this definition, a standard risk matrix has been built, as shown below:
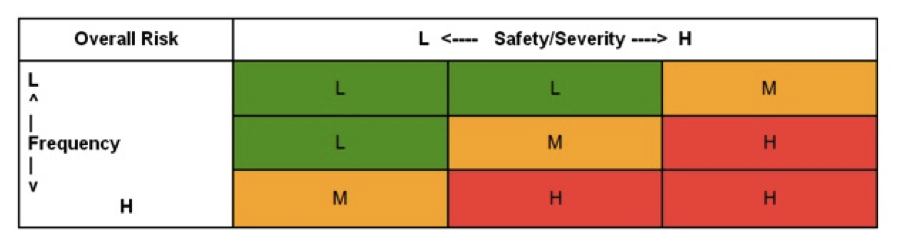 * Sample matrix – your organization MUST develop (and justify) your own criteria.
* Sample matrix – your organization MUST develop (and justify) your own criteria.
Where the horizontal columns are for safety/severity/quality, and the vertical rows represent probability/frequency/detectability. After developing the acceptance criteria the process proceeds as follows:
- Find the system function categories and add them to a risk assessment table. These categories are determined by reviewing the URS and assessing how each of the requirements correlates to a similar system function (grouping).
- Determine the risk associated with each URS function in terms of potential failure or if the function/system is not available (off-line or non-functioning).
- Determine Severity/Safety/Quality impact on the associated failure.
- Determine the Frequency/Probability/Detectability of the possibility of failure.
- Utilize the acceptance criteria chart to find overall risk.
- Place the overall risk into one of the three risk classes (High/Medium/Low).
- Complete the same risk assessment on each of the functional items listed in the URS.
VALIDATION PRIORITY LEVEL
Once risk assessments for individual functional items from the URS have been determined, a validation approach for each functional category can be assembled. The following best practice approach outlines three types of validations that can be utilized with a risk based process.
- High: Complete/comprehensive testing required. All system and sub-systems must be thoroughly tested according to a scientific, data driven rationale. This is similar to the ‘classic’ approach to validation. Furthermore, enhancement may be needed to improve detectability of failure via in-process production controls.
- Medium: Testing of the functional requirements per the URS/FRS required with sufficient assurance that the item has been properly characterized.
- Low: No formal testing needed, but presence (detectability) of the functional item may be required.
These criteria are then applied to the table of functional items and the output is the validation test plan, as described in the section below.
VALIDATION PLAN
The new guidances for process validation have established that “the collection and evaluation of data, from the process design stage through production, which establishes scientific evidence that a process is capable of consistently delivering quality products”. This has resulted in validation being split into three stages:
- Stage 1: Process Design – The commercial process based on experience gained from development and scale-up.
- Stage 2: Process Qualification – the reproducible, commercial scale is confirmed on the basis of process design.
- Stage 3: Continued Process Verification – To show that the process is in a state of control during routine production.
Manufacturers must be able to prove with a high degree of assurance that the product can be manufactured according to the quality attributes before a batch is placed on the market. For this purpose, data from a lab, scale-up and pilot scale are meant to be used. The data are explicitly meant to cover conditions involving a great range of process variation.
The manufacturer must:
- Determine and understand the process variations.
- Detect these process variations and assess their extent.
- Understand the influence on the process and the product.
- Control such variations depending on the risk they represent.
Qualification activities lacking the basis of a sound process understanding will not lead to an accordingly safe product. The process must be maintained during routine operations, including materials, equipment, the environment, personnel and changes in the manufacturing procedures.
PROCESS DESIGN STAGE
This stage is about building and capturing process knowledge and understanding; the manufacturing process is meant to be defined and tested, which will then be reflected in the manufacturing and testing documentation. Earlier development stages do not have to be conducted under cGMP. The basis should be sound scientific methods and principles, including Good Documentation Practice. There is no regulatory expectation that the process be developed and tested until it fails, but a combination of conditions involving a high process risk should be known. In order to achieve this level of process understanding, the implementation of Design of Experiments (DOE) in connection with risk analysis tools are recommended. Other methods, such as classical laboratory tests, are also considered acceptable. Adequate documentation of the process understanding based on rationale is essential. Establishing a strategy for process control and understanding are considered to be the basis for this stage.
PROCESS QUALIFICATION STAGE
This stage proves that the process design is suitable for reproducibly manufacturing commercial batches. This stage contains two steps: qualification activities regarding facilities and equipment, and performance qualification (PQ). This stage encompasses the activities that are currently summarized under process validation; qualified equipment is used to demonstrate the process can create a product in conformity with the specifications. The terms Design Qualification (DQ), Installation Qualification (IQ) and Operational Qualification (OQ) are no longer described as part of the qualification. They are, however, still conceptually used within the validation plan. The plan should cover the following items:
- Test Description.
- Acceptance criteria.
- Schedule of Validations.
- Responsibilities.
- Protocol with Pre-Approval / Post Approval.
- Change Control.
- Results.
CONTINUED VALIDATION STAGE
The final stage is to keep the validated state of the process current during routine production. The manufacturer is required to establish a system to detect unplanned process variations. Data are meant to be evaluated accordingly (in-process) so that the process does not get out of control. The data must be statistically trended, and the analysis be done by a qualified person. These evaluations are meant to be reviewed by the quality unit in order to detect changes in the process (alert limits) at an early stage and to be able to implement process improvements. Unexpected process changes can also occur, even in a well-developed process. Here, the guidance recommends “that the manufacturer use quantitative, statistical methods whenever feasible” in order to identify and investigate for root cause. At the beginning of routine production, the guidance recommends the same scope and frequency of monitoring activities and sampling occurs as in the process qualification stage until enough data has been collected.
Analysis from complaints, OOS results, deviations and non-conformances can also give data/trends regarding process variability. Employees on the production line and in quality assurance should be encouraged to give feedback on the process performance. Operator errors should also be tracked in order to check if training measures are appropriate.
Finally, these data sets can be used to develop process improvements. The changes however, may only be implemented in a structured way and with the final approval by quality assurance and potential re-validation in the process qualification stage.
Reprinted with permission from MasterControl Inc. and GxP Lifeline”
Bio:
Peter Knauer is a partner consultant with MasterControl’s Quality and Compliance Advisory Services. He has more than 20 years of international experience in the biomedical industry, primarily focusing on supply chain management, risk management, CAPA, audits and compliance issues related to biopharmaceutical and medical device chemistry, manufacturing and controls (CMC) operations. He was most recently head of CMC operations for British Technology Group in the United Kingdom and he has held leadership positions for Protherics UK Limited and MacroMed. Peter started his career at Genentech, where he held numerous positions in engineering and manufacturing management. Peter is currently chairman of the board for Intermountain Biomedical Association (IBA) and a member of the Parenteral Drug Association (PDA). Peter holds a master’s degree in biomechanics engineering from San Francisco State University and a bachelor’s degree in materials science engineering from the University of Utah. Contact him at
pknauer@mastercontrol.com.